了解爱思益普最新资讯、活动动态
News Detail
 来源:2024-11-20 15:18:16
来源:2024-11-20 15:18:16
 浏览量:13512
浏览量:13512

自2001年以来,蛋白激酶抑制剂格列卫(伊马替尼)是第一个获得FDA批准的药物,标志着针对BCR-ABL癌症治疗的重大突破,并预示着蛋白激酶抑制剂在肿瘤学和其他治疗领域的兴起。在过去的二十多年中,FDA共批准多达88种小分子抑制剂(截止到2024年8月30日),其中靶向蛋白激酶83种,靶向脂质激酶5种,详见图3。大多数获批的小分子激酶抑制剂通过结合目标的ATP结合位点来抑制蛋白激酶的活性。此外,变构抑制剂和共价抑制剂也是重要的发展方向,它们通过模拟蛋白激酶的自然调控机制,提供高特异性和治疗潜力。根据早期报告,我们将小分子蛋白激酶抑制剂分为七个主要类别:
I型:这些抑制剂可以与激活环中的氨基酸残基形成氢键,通常与ATP竞争性结合。
I?型:I?型抑制剂与ATP结合位点结合,但与I型抑制剂相比,与激活环的相互作用较弱。
II型:II型抑制剂与激酶的DFG-out构象结合,这通常在激酶的非活跃状态下观察到。这些抑制剂可以稳定这种非活跃状态,从而抑制激酶活性。
我们进一步将I?型和II型拮抗剂细分为A和B两个亚型。A型药物延伸过守门残基进入背部凹槽。相反,B型药物不延伸进入背部凹槽。根据不完整的数据表明,这种差异的可能意义是A型拮抗剂与其酶靶标结合的停留时间比B型阻断剂更长。

图1. Type I和TypeII结合位点
III型和IV型:变构抑制剂也是蛋白激酶抑制剂开发中的一个重要方向。根据变构抑制剂的结合位点是否靠近激酶的ATP结合位点,它们可以被分为III型和IV型抑制剂。III型抑制剂的结合位点靠近激酶的ATP结合位点,所有经过临床验证的MEK1/MEK2抑制剂都属于这一类。IV型抑制剂的结合位点则离ATP结合位点更远,包括ABL抑制剂如ABL001和AKT1抑制剂如MK-2206。
V型:V型抑制剂是针对性共价不可逆抑制剂,它们与激酶的特定氨基酸残基形成共价键,导致激酶永久性失活。
VI型:针对激酶的假激酶域。假激酶约占所有人类激酶的10%,被定义为缺乏一个或多个必需的保守催化基序的激酶,因此被预测为催化活性不活跃。尽管缺乏催化活性,假激酶的突变或其表达的失调已被与多种疾病联系起来,尤其是癌症和自身免疫疾病。因此,假激酶被认为是一类重要的新兴药物靶标。
Type VII型:靶向激酶的胞外结构域。

图2. Type III, IV, VI, VII抑制剂结合位点示意图
小分子蛋白激酶抑制剂(PKIs)的未来前景是充满希望的。随着新药的不断开发,大约有110种新型激酶处于临床试验阶段,这表明这一领域仍有很大的未开发潜力。未来的研究将集中在克服药物抗性、优化药代动力学和药效学特性以及个性化治疗上。联合疗法也将成为提高疗效的重要策略,通过将PKIs与其他治疗方法结合,可能有效地解决药物抗性问题。

图3. FDA批准的蛋白激酶抑制剂
ICE拥有用于筛选各种类型抑制剂的蛋白质,并且提供相应的筛选服务。进入下面的链接或通过我们的网站发送消息来了解更多信息:https://protein.ice-biosci.com/
ABL抑制剂:
在2003年,Kuriyan和Superti-Furga的研究小组报告称,肉豆蔻酰基团参与了ABL1和ABL2激酶的自我调节:通过与ABL1激酶域C-叶中的内部“肉豆蔻酰结合位点/口袋”结合,共价连接到ABL1 N-末端的肉豆蔻酰基团,诱导了一个组装的非活跃状态,即SH1,SH2,SH3自抑单元。这种在ABL1中观察到的自我抑制机制可以与BCR片段融合后丧失,BCR片段取代了包含肉豆蔻酰化位点的ABL1的N-末端帽区域,从而导致BCR-ABL1的组成性激活。这种组装的非活跃状态与II型抑制剂如Imatinib和Olverembatinib不同,他们是通过直接结合DFG-out环构象来稳定激酶域的非活跃构象。通过与肉豆蔻酰位点相互作用来抑制ABL1激酶的药物将比ATP竞争性酪氨酸激酶抑制剂(TKIs)更具选择性,并且不会受到由于非靶激酶抑制而引起的不良事件的影响,因此可能具有良好的耐受性。

图4. ABL非活性状态下的最小自抑结构
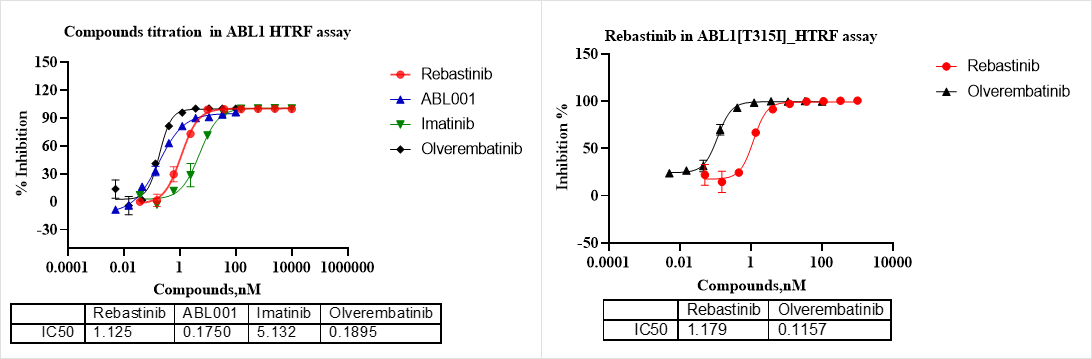
图5. Rebastinib, Imatinib 和 Olverembatinib 为ABL II型抑制剂,ABL001为ABL type IV型抑制剂。
MEK1抑制剂:
RAF/MEK/ERK通路是控制细胞生理的核心,其失调与许多癌症有关。因此,构成这一途径的蛋白一直受到强烈的药物发现和开发努力的影响。变构MEK抑制剂(Allosteric MEK inhibitors, MEKi)对RAF/ MEK/ERK通路信号通路具有复杂的作用,在临床上与BRAF抑制剂联合应用于恶性黑色素瘤。变构MEKi可以通过至少四种不同机制中的一种或多种来阻断RAF/MEK/ERK信号通路的激活:1)通过与uMEK结合并诱导阻止其与RAF结合;2)允许uMEK与RAF结合,使MEK稳定于抗磷酸化构象;3)通过阻止RAF释放pMEK,阻止ERK磷酸化;4)抑制游离pMEK以阻断其磷酸化ERK的能力。研究结果表明,MEK抑制剂除抑制MEK外还对BRAF/MEK复合物起抑制作用。

图6. MEK1变构抑制剂结合位点

图7. Selumetinib, Trametinib,和Avutometinibe 是III 型MEK1抑制剂
EGFR抑制剂:
吉非替尼、厄洛替尼、阿法替尼和奥西替尼已被批准用于治疗EGFR激酶中存在激活突变的非小细胞肺癌,但由于迅速产生的耐药性,最常见的是由T790M突变产生许多不良反应。而EAI045不直接与EGFR的ATP结合位点竞争,而是结合到EGFR激酶域的“C-螺旋外”(C-helix out)构象的变构位点。在EGFR的不对称二聚体中,激活子亚基的C-叶与接收子亚基的N-叶相互作用,导致接收子亚基的C-螺旋向内移动,形成活跃构象。EAI045通过稳定C-螺旋在外的位置,阻止这种构象变化,从而抑制EGFR的活性。在突变型EGFR中,由于突变导致的构象变化,二聚体的形成可能更加频繁。EAI045通过阻断这种二聚体相互作用,减少了EGFR的激活。由于EAI045的高度选择性,它不太可能抑制其他非靶标激酶,这可能减少了因非特异性抑制而产生的不良反应。因此,EAI045提供了一种新的治疗策略,尤其适用于那些对传统ATP竞争性酪氨酸激酶抑制剂产生耐药的癌症患者,有望改善治疗效果并减少副作用。

图8. 变构型EGFR抑制剂的结构与结合方式

图9. Afatinib和Osimertinib 为EGFR共价抑制剂。BLU-945 为I型抑制剂,EAI-045是III抑制剂。
AKT抑制剂:
AKT家族蛋白在细胞信号传导中扮演着核心角色,它们参与调节细胞增殖、存活、生长、代谢和血管生成等关键生物学过程。AKT属于AGC超家族,具有N端的PH结构域、激酶结构域和C端的调节区域。在生长因子的刺激下,PI3K的激活导致AKT转移到质膜上,并在T308和S473两个位点发生磷酸化,这一过程分别由PDK1和mTORC2催化,是AKT激活的关键步骤。在多种癌症中,AKT的激活常常是由于上游基因如PIK3CA的突变或扩增,以及PTEN的失活或丢失。此外,AKT基因本身的突变,如AKT1的E17K突变,也能导致其病理性激活,这种突变改变了AKT的脂质结合特异性,导致其在细胞膜上的异常定位和持续的信号传导。
最新的研究表明,在没有上游信号的情况下,AKT的PH结构域和激酶结构域之间的相互作用使其保持在一种自抑制的闭合状态,这阻止了PDK1对T308位点的磷酸化。而当接收到上游信号时,AKT会从这种自抑制状态转变为开放状态,使得磷酸化和激活成为可能。研究者通过对大量人类肿瘤样本的分析发现,PH-KD接触位点的突变在癌症中是存在的,这种突变破坏了PH和KD之间的相互作用,可能是AKT在癌症中激活的一种新机制。更重要的是,这些肿瘤特异性的突变不仅能够激活AKT,还可能改变肿瘤对变构AKT抑制剂的敏感性,这对于开发新的癌症治疗策略具有重要意义。

图10. ATK两种不同激活方式

图11. ATK变构抑制剂结合位点

图12. Capivasertib 为I 型AKT抑制剂,MK2206为IV AKT 型AKT抑制剂
JAK抑制剂

图13. BMS-986165与TYK2 JH2共晶结构
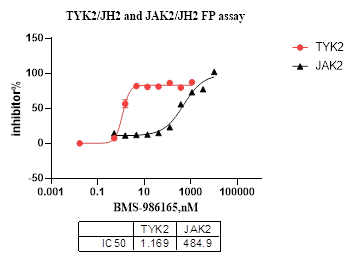
图14. BMS-986165为TYK2 VI 型抑制剂
BTK抑制剂:
共价 BTK 抑制剂与 BTK 的 ATP 结合口袋内的半胱氨酸481(C481)残基形成不可逆的共价键。BTK 抑制导致 PLCγ2 等主要下游信号通路的激活受损,阻止恶性 B 细胞增殖并促进细胞凋亡。此类已批准的抑制剂包括伊布替尼、阿卡替尼和泽布替尼。临床前分析显示,共价抑制剂对 C481S 突变细胞几乎没有活性,对获得性耐药性的患者构成潜在挑战 。新的可逆BTK 抑制剂旨在最大限度地减少副作用和减少耐药性。Pirtobrutinib是目前FDA批准的第一个也是唯一一个非共价、可逆的BTK抑制剂。LP-168能够通过共价结合抑制野生型BTK和通过非共价结合抑制耐药突变型BTK,从而抑制B细胞受体信号传导通路,阻断B淋巴细胞的异常增殖、分化和存活。另一种针对 BTK 的新兴治疗选择是使用 BTK 降解剂NX-2127 目前正在复发或难治性 B 细胞恶性肿瘤中进行 1 期临床试验。
尽管大多数CLL (chronic lymphocytic leukemia) 患者对单药治疗 BTK 抑制剂有多年的反应,但需要持续治疗,并且最终会发展出 BTK 或 PLCγ2 的耐药突变。共价抑制剂特别容易受到 C481 突变的影响,因为它们依赖于与该残基的结合。非共价 BTK 抑制剂克服了这一限制,但仍易受到新型 BTK 激酶结构域突变的影响。BTK L528W、V416L 和 A428D 被归类为激酶功能受损的耐药突变,而 BTK C481S 和 T474I 被归类为激酶功能完整突变。BTK 降解剂仍在进行临床试验,但早期结果表明它们可以克服激酶受损和激酶熟练的获得性 BTK 耐药突变,但尚不清楚它们是否可以靶向下游 PLCγ2 激活突变。

图15. FDA批准的BTK抑制剂
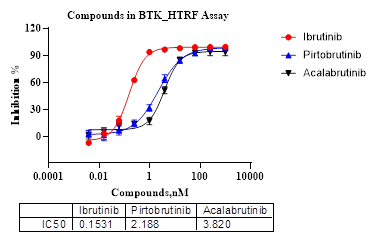
图16. Ibrutinib和Acalabrutinib 为BTK共价抑制剂,Pirtobrutinib为BTK I型抑制剂
参考文献
1. Lu X, Smaill JB, Ding K. New Promise and Opportunities for Allosteric Kinase Inhibitors. Angew Chem Int Ed Engl. 2020 Aug 10;59(33):13764-13776.
2. Cohen P, Cross D, J?nne PA. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov. 2021 Jul;20(7):551-569.
3. Attwood MM, Fabbro D, Sokolov AV, Knapp S, Schi?th HB. Trends in kinase drug discovery: targets, indications and inhibitor design. Nat Rev Drug Discov. 2021 Nov;20(11):839-861.
4. Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2024 update. Pharmacol Res. 2024 Feb;200:107059.
5. Schoepfer J, Jahnke W, Berellini G, Buonamici S, Cotesta S, Cowan-Jacob SW, Dodd S, Drueckes P, Fabbro D, Gabriel T, Groell JM, Grotzfeld RM, Hassan AQ, Henry C, Iyer V, Jones D, Lombardo F, Loo A, Manley PW, Pellé X, Rummel G, Salem B, Warmuth M, Wylie AA, Zoller T, Marzinzik AL, Furet P. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J Med Chem. 2018 Sep 27;61(18):8120-8135.
6. Chan WW, Wise SC, Kaufman MD, Ahn YM, Ensinger CL, Haack T, Hood MM, Jones J, Lord JW, Lu WP, Miller D, Patt WC, Smith BD, Petillo PA, Rutkoski TJ, Telikepalli H, Vogeti L, Yao T, Chun L, Clark R, Evangelista P, Gavrilescu LC, Lazarides K, Zaleskas VM, Stewart LJ, Van Etten RA, Flynn DL. Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036. Cancer Cell. 2011 Apr 12;19(4):556-68.
7. Gonzalez-Del Pino GL, Li K, Park E, Schmoker AM, Ha BH, Eck MJ. Allosteric MEK inhibitors act on BRAF/MEK complexes to block MEK activation. Proc Natl Acad Sci U S A. 2021 Sep 7;118(36):e2107207118.
8. Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, Xu C, Rhee K, Chen T, Zhang H, Palakurthi S, Jang J, Lelais G, DiDonato M, Bursulaya B, Michellys PY, Epple R, Marsilje TH, McNeill M, Lu W, Harris J, Bender S, Wong KK, J?nne PA, Eck MJ. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016 Jun 2;534(7605):129-32.
9. Parikh C, Janakiraman V, Wu WI, Foo CK, Kljavin NM, Chaudhuri S, Stawiski E, Lee B, Lin J, Li H, Lorenzo MN, Yuan W, Guillory J, Jackson M, Rondon J, Franke Y, Bowman KK, Sagolla M, Stinson J, Wu TD, Wu J, Stokoe D, Stern HM, Brandhuber BJ, Lin K, Skelton NJ, Seshagiri S. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc Natl Acad Sci U S A. 2012 Nov 20;109(47):19368-73.
10. Bae H, Viennet T, Park E, Chu N, Salguero A, Eck MJ, Arthanari H, Cole PA. PH domain-mediated autoinhibition and oncogenic activation of Akt. Elife. 2022 Aug 15;11:e80148.
11. Wrobleski ST, Moslin R, Lin S, Zhang Y, Spergel S, Kempson J, Tokarski JS, Strnad J, Zupa-Fernandez A, Cheng L, Shuster D, Gillooly K, Yang X, Heimrich E, McIntyre KW, Chaudhry C, Khan J, Ruzanov M, Tredup J, Mulligan D, Xie D, Sun H, Huang C, D'Arienzo C, Aranibar N, Chiney M, Chimalakonda A, Pitts WJ, Lombardo L, Carter PH, Burke JR, Weinstein DS. Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165. J Med Chem. 2019 Oct 24;62(20):8973-8995.
12. Cool A, Nong T, Montoya S, Taylor J. BTK inhibitors: past, present, and future. Trends Pharmacol Sci. 2024 Aug;45(8):691-707.


业务咨询
北京
业务咨询专线:010-6780-9840
联系地址:北京市经济技术开发区科创十三街18号院锋创科技园16号楼
上海
业务咨询专线:010-6780-9840
联系地址:上海市浦东新区蔡伦路780号新技术推广大楼3E5O室
徐州
业务咨询专线:010-6780-9840
联系地址:江苏省徐州市云龙区淮海文博园 二号楼2层
贵州
业务咨询专线:010-6780-9840
联系地址:贵州省贵阳市南明区龙岭路50号 欧美医药产业园一期2号楼












