了解爱思益普最新资讯、活动动态
News Detail
 来源:本站
来源:本站
 浏览量:18708
浏览量:18708

乳腺癌是全世界癌症相关死亡的主要原因之一,也是导致女性死亡最常见的癌症类型。2023年11 月 16 日,美国 FDA宣布批准阿斯利康 Capivasertib上市,其为首个批准的AKT抑制剂,与Faslodex(氟维司群)联用,用于治疗激素受体阳性(HR+)、人表皮生长因子受体2阴性(HER2-)局部晚期或转移性乳腺癌患者。由于较早布局AKT抑制剂的公司如默沙东(MK-2206)、礼来(LY2780301)等均以失败告终,本土药企开始将目光投向这个横跨多个大癌种的潜力靶点。

AKT是一种丝氨酸/苏氨酸蛋白激酶,在代谢和癌症中扮演着核心角色。AKT家族由三个成员组成,即AKT1、AKT2和AKT3。它们属于蛋白激酶A、激酶G和激酶C(AGC)超家族的丝氨酸/苏氨酸激酶,参与调节包括细胞增殖、存活、生长、代谢和血管生成在内的关键细胞过程。AKTs包括一个N端磷脂酰肌醇结合(PH)结构域、一个激酶结构域(KD)和一个含有疏水基序的C端调节区域。3种亚型的 PH 结构域包 含110 个氨基酸,其中大约60%是相同的,铰链区大致有25%是相同的,而激酶区有90%是同源的。3 种亚型的ATP 结合位点的氨基酸残基是相同的。

图1. AKT家族成员及突变类型
AKT是一种具有高度灵活性的蛋白质,它在细胞质中以一种动态平衡的形式存在,这种平衡由其PH结构域调节。当细胞接收到生长因子的信号时,PI3K被激活,并将PIP2磷酸化,生成PIP3。PIP3作为信号分子,能够与AKT的PH结构域结合,这一过程促使AKT从细胞质转移到细胞膜。在细胞膜上,AKT的Thr308位点被PDK1磷酸化。活化的AKT随后触发下游信号分子的级联反应,进而调控细胞的多种生命活动。

图2. AKT激活机制
此外,Bae, Viennet et al.报道R86A PH结构对激酶结构域亲和力(KD 107 μM)比WT PH结构域对激酶结构域亲和力(KD 37 μM)大约高三倍,表明R86A突变通常可以增强PH结构域和激酶结构域之间的自抑制相互作用。但在存在AKT变构抑制剂MK2206的情况下使用MST测量R86A PH结构域对激酶结构域的亲和力,但未能检测到可检测的结合(KD > 250 μM),可能为MK2206诱导的PH结构域和激酶结构域的相互作用与R86A的效果不同。

图3. AKT激酶结构与与PH结构域示意图。紫色为PH结构域,黄色为激酶结构域,灰色为变构抑制剂结合位点
PH结构域突变体AKT[E17K]是一个已经确立的致癌基因。PH结构域中的Glu17和Tyr18及激酶结构域中的Phe309在AKT自抑制中也发挥关键作用。Tyr18和Phe309在AKT形式中是高度保守的残基。Tyr18与AKT激酶结构域的催化核心非常接近, Tyr18-Phe309结构域间相互作用可能阻止Akt底物获得有利于磷酸转移反应的有利取向,从而自抑制AKT的酶活性。

图4. PH结构域Tyr18和Phe309在Akt激酶结构域激活环上的相互作用示意图
AKT蛋白具有多个潜在的结合位点,这些位点的占据可以抑制AKT的特定功能,进而对信号传导路径产生影响。这些结合位点主要包括:PH结构域上的PIP3结合位点、C端磷酸化位点附近的疏水口袋以及ATP结合位点。除此之外,PH结构域、铰链区和激酶区之间的相互作用和构象变化也暴露了一些分子间结合位点。基于这些不同的结合位点,AKT抑制剂可以被分类为以下几种类型:针对PH结构域的抑制剂、与ATP竞争结合的抑制剂以及变构抑制剂。

图5. Capivasertib在AKT上抑制作用
MK-2206是一种变构抑制剂,需要完整的PH-KD界面才能发挥作用。MK-2206在体外对AKT同工酶1、2和3的抑制作用的IC50值分别为8、12和65nM。MK-2206还可以通过抑制T308和S473的自磷酸化,阻止AKT介导的下游信号分子的磷酸化,包括TSC2、PRAS40和核糖体S6蛋白。当MK-2206与不同药物联合使用时,可观察到协同作用。例如MK-2206与EGFR抑制剂厄洛替尼联合使用用于非小细胞肺癌细胞系,或与双重EGFR-Her2抑制剂拉帕替尼联合使用用于乳腺癌细胞系。

图6. MK-2206分子结构
最近加州大学Gregory B. Craven等人报道了一种专门针对 PH 结构域中的 Glu17 (E17K)突变的共价抑制剂compound4,化合物4含有一个水杨醛基团,这个基团能够与AKT1 E17K突变体中的特定氨基酸残基(Lys17)形成共价键。化合物4与AKT1 E17K突变体结合后,能够在细胞内招募内源性的Zn2+离子,并与Cys296和Cys310形成一个新的锌离子螯合物。Zn2+离子的加入进一步稳定了化合物4与AKT1 E17K突变体的复合物,增强了其热稳定性和靶标结合的持久性。另外,化合物4通过稳定AKT1 E17K突变体的自抑制状态,阻止Thr308和Ser473位点的自磷酸化,从而抑制AKT的下游信号传导。

图7. Zn2+螯合在AKT1(E17K)抑制中的作用
AKT除在肿瘤领域有较大的研究意义外,在代谢中也起到关键作用。在代谢综合征(MetS)中,AKT(蛋白激酶B)是一个关键的调节因子,通过胰岛素信号通路发挥着至关重要的作用。胰岛素与胰岛素受体结合,激活IRS,进而激活PI3K/AKT通路,这对于维持胰岛素抵抗(IR)至关重要。AKT的下游效应包括促进GLUT4的转位、增强胰岛素分泌、抑制肝细胞中的糖异生以及激活SREBP1c以促进脂质合成。此外,AKT信号还调节内皮功能、抑制自噬,并可能通过增加活性氧(ROS)水平影响细胞功能。总之,AKT在调节糖代谢、脂质代谢和内皮功能中发挥着核心作用,影响着代谢综合征的发生和发展。综上所述,AKT是一个有较大潜力的靶点。
爱思益普打造了集蛋白纯化、酶学方法开发、抑制剂活性筛选于一体的AKT筛选平台。同时协同细胞、ADME等平台,可支持AKT药物活性评价的一体化研究,旨在建立全面的靶点筛选和体外生物学研究平台,满足新药研发企业对速度、效率和结果的需求,用专业的技术和高效的沟通帮助客户提高新药研发的效率。
1. AKT Biochemical Assay

图8. Capivasertib和MK-2206对AKT抑制作用
2. LC-MS characterization for covalent inhibitor of AKT
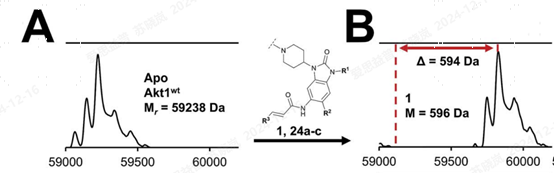
图9. LC-MS平台可供AKT共价抑制剂筛选
3. AKT NanoBRET TE assay
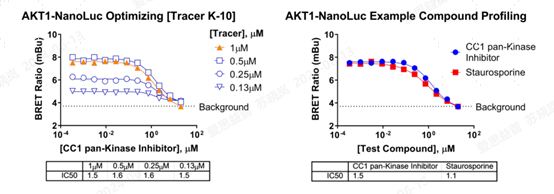

图10.不同AKT抑制剂对 NanoBRET TE assay的抑制作用
4. Cell proliferation Assay

图11.不同AKT抑制剂对细胞系的增殖抑制作用
5. Biomarker detection_ phosporylation level detected by HTRF,WB,ICW

图12.下游Biomarker pS6、GSK3β和pAKT检测结果
6. In vitro glucose uptake assay

7. BaF3-AKT1-E17K CDX model

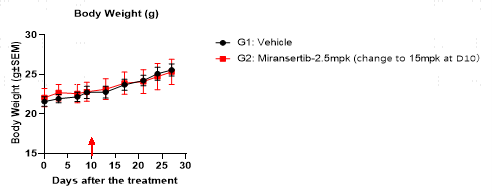
图13. 在注射了BaF3-AKT1-E17K肿瘤细胞的BALB/c裸鼠中进行的肿瘤生长和体重变化实验。比较对照组与米拉尼司替布(Miransertib)治疗组,该治疗组最初以2.5毫克/千克的剂量给药,并在治疗10天后增加到15毫克/千克。研究结果表明,米拉尼司替布对肿瘤生长有显著影响,而对体重的影响最小。
8. Kinase panel/Profiling

图14.以FGFR 抑制剂Infigratinib为例
参考文献
1. Li Yan, et al. Abstract #DDT01-1: MK-2206: A potent oral allosteric AKT inhibitor. 2009.
2. Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, Li J, Gao B, Ji Q, Maynard J, Ricketts SA, Cross D, Cosulich S, Chresta CC, Page K, Yates J, Lane C, Watson R, Luke R, Ogilvie D, Pass M. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012 Apr;11(4):873-87.
3. Craven GB, Chu H, Sun JD, Carelli JD, Coyne B, Chen H, Chen Y, Ma X, Das S, Kong W, Zajdlik AD, Yang KS, Reisberg SH, Thompson PA, Lipford JR, Taunton J. Mutant-selective AKT inhibition through lysine targeting and neo-zinc chelation. Nature. 2024 Nov 6.
4. Parikh C, Janakiraman V, Wu WI, Foo CK, Kljavin NM, Chaudhuri S, Stawiski E, Lee B, Lin J, Li H, Lorenzo MN, Yuan W, Guillory J, Jackson M, Rondon J, Franke Y, Bowman KK, Sagolla M, Stinson J, Wu TD, Wu J, Stokoe D, Stern HM, Brandhuber BJ, Lin K, Skelton NJ, Seshagiri S. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc Natl Acad Sci U S A. 2012 Nov 20;109(47):19368-73.
5. Bae H, Viennet T, Park E, Chu N, Salguero A, Eck MJ, Arthanari H, Cole PA. PH domain-mediated autoinhibition and oncogenic activation of Akt. Elife. 2022 Aug 15;11:e80148.


业务咨询
北京
业务咨询专线:010-6780-9840
联系地址:北京市经济技术开发区科创十三街18号院锋创科技园16号楼
上海
业务咨询专线:010-6780-9840
联系地址:上海市浦东新区蔡伦路780号新技术推广大楼3E5O室
徐州
业务咨询专线:010-6780-9840
联系地址:江苏省徐州市云龙区淮海文博园 二号楼2层
贵州
业务咨询专线:010-6780-9840
联系地址:贵州省贵阳市南明区龙岭路50号 欧美医药产业园一期2号楼












