了解爱思益普最新资讯、活动动态
News Detail
 来源:2024-12-24 15:08:58
来源:2024-12-24 15:08:58
 浏览量:15600
浏览量:15600

成纤维细胞生长因子受体(Fibroblast growth factor receptor, FGFR)是在细胞膜上表达的受体酪氨酸激酶家族,人成纤维细胞生长因子受体(FGFR)家族由四个成员组成:FGFR1 至 FGFR4。FGFR与其他受体酪氨酸激酶(RTK)类似,可被细胞外信号刺激和激活。FGFR的天然配体是成纤维细胞生长因子(FGF),FGF的结合驱动FGFR 的二聚化,诱导细胞内激酶结构域的自磷酸化,然后激活下游转导途径,在细胞增殖、分化、迁移和存活中都起着至关重要的作用。FGFR失调与多种癌症有关,例如尿路上皮癌、肝细胞癌、卵巢癌和肺腺癌,FGFR被认为是治疗各种癌症的药物靶点。
1、爱思益普已表达纯化FGFR系列活性蛋白可供选择

图1. ICE已构建的FGFR家族及其突变蛋白的生化实验
2、蛋白产品案例展示:FGFR3[V555M]
A:生产流程:
FGFR3[V555M]在HEP sf9 高表达昆虫细胞中进行表达。

图2. FGFR3[V555M]生产流程图
B:常规QC结果:
SDS-PAGE检测FGFR3[V555M]的纯度在95%以上。

图3. FGFR3[V555M] SDS-PAGE结果图
C:活性验证结果:
HTRF法对FGFR3[V555M]蛋白分别进行酶滴和阳性药的验证,FGFR3[V555M]具有良好的活性,满足实验需求。
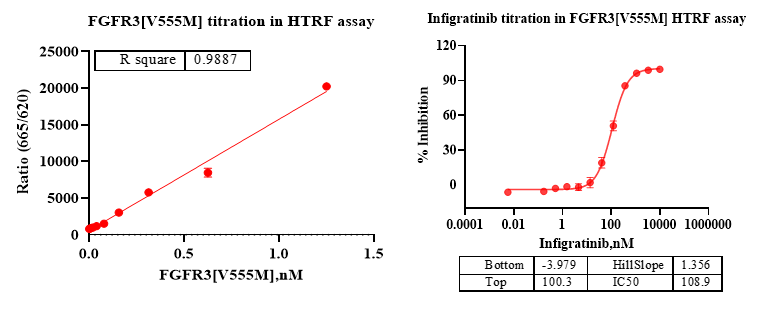
图4. HTRF法活性测定
3、不同抑制剂对FGFR家族IC50检测数据
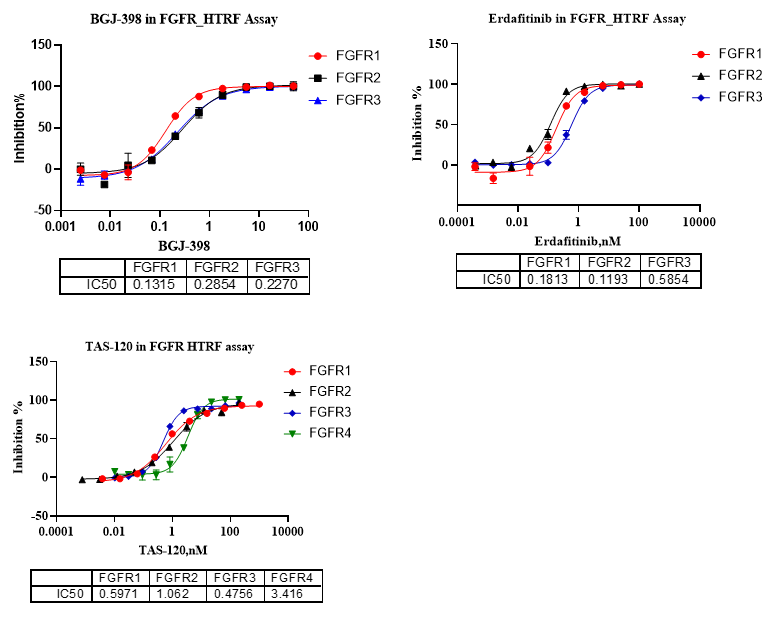
图5. HTRF法检测不同抑制剂对FGFR家族的IC50
4、爱思益普FGFR系列蛋白产品列表

除了丰富的现货产品,同时可以提供克隆-表达-纯化-质控-活性测定一体化蛋白定制服务。以满足您对蛋白序列、标签或修饰以及不同应用场景的个性化需求。
5、FGFR 背景介绍
成纤维细胞生长因子(FGF)由保守的核心域组成,可以是球状或非典型的β-三叶草结构,可以分为canonical FGFs(FGF1-10、FGF16-18、FGF20和FGF22)、内分泌FGFs(FGF19/FGF15、FGF21和FGF23)以及FGF同源因子(FGF11-14)。全长的FGF受体(FGFR1-4)是具有三个细胞外免疫球蛋白样结构域和一个细胞内酪氨酸激酶结构域的I型跨膜蛋白。Canonical FGFs与肝素硫酸共因子一起通过FGFR传递信号,而内分泌FGFs与klotho共受体(KLA和KLB)和肝素硫酸共因子一起通过FGFR传递信号。成纤维细胞生长因子受体(FGFR)信号传导在细胞生理和病理过程中扮演着关键角色,尤其是在肿瘤的发生和发展中。
FGFR发生二聚化,激活其内在的酪氨酸激酶活性,导致受体自身酪氨酸残基的磷酸化,即自磷酸化。FGFR信号传导受酪氨酸激酶域中四个保守的酪氨酸残基的自磷酸化和c端区域一个被ERK磷酸化的丝氨酸残基调控。在FGFR1中,激活环(A环)中的Y653和Y654参与FGFR激酶的激活;αH螺旋中的Y730参与ERK和PI3K信号的激活;激酶尾部的Y766参与磷脂酶Cγ (PLCγ)的募集和激活;c端尾部的S777参与负反馈调节。自磷酸化的FGFR进一步激活多条下游信号通路,包括:
a)PI3K-AKT通路:该通路在细胞存活、增殖和代谢中起关键作用,通过增加细胞内PI3K的活性,进而激活AKT,促进细胞的生长和存活。
b)RAS-ERK通路:该通路涉及细胞增殖、分化和迁移,通过激活RAS蛋白,进而激活RAF、MEK和ERK激酶,影响细胞周期的进程。
c)PLCγ通路:该通路通过激活磷脂酶Cγ(PLCγ),导致细胞内钙离子浓度变化和蛋白激酶C(PKC)的激活,影响细胞的迁移和增殖。
异常激活的FGFR信号通路与多种癌症的发生发展有关。这种异常激活可能由基因融合、基因扩增、基因突变或替代信号通路的激活引起,导致肿瘤细胞的增殖和存活。

图6. FGFR家族成员及磷酸化位点示意图

图7. FGFR信号通路
目前已经开发了多种小分子FGFR抑制剂,用于治疗FGFR相关的癌症患者,例如尿路上皮癌、脑癌、乳腺癌、胆管癌(CCA)、胃癌、肝癌、肺癌以及卵巢癌和子宫癌。与酪氨酸激酶域ATP结合位点结合的抑制剂通常被分类为多激酶FGFR抑制剂(第一代FGFR抑制剂)或选择性FGFR抑制剂(第二代FGFR抑制剂)。Derazantinib、dovitinib、tasurgratinib、lenvatinib、lucitanib、nintedanib和ponatinib都是多激酶FGFR抑制剂,除抑制FGFR之外,还对多个其他RTKs有活性,可能由于FGFR对其他RTKs在血管生成和免疫调节以及肿瘤发生中有作用,而潜在地靶向癌细胞及其微环境。由于它们的广泛活性,所以细胞毒性也会较大。
选择性FGFR抑制剂包括泛FGFR、FGFR1/2/3和FGFR2/3抑制剂,以及选择性FGFR2、FGFR3或FGFR4抑制剂。Lirafugratinib是一种选择性FGFR2抑制剂,LOXO-435是一种选择性FGFR3抑制剂,roblitinib和fisogatinib是选择性FGFR4抑制剂。FGFR抑制剂也可以根据药物-靶标相互作用的机制被分类为非共价或共价抑制剂。非共价FGFR抑制剂,如erdafitinib、pemigatinib、infigratinib、rogaratinib和zoligratinib,以ATP竞争性方式抑制FGFR的酶活性。Erdafitinib已被批准用于治疗FGFR3改变的mUC,但该药与多种不可耐受的靶向毒性有关,易发生与耐药性相关的FGFR3门卫(gatekeeper)突变,临床应用受到一定限制。共价FGFR抑制剂与ATP结合口袋中特定的半胱氨酸残基相互作用,具有更高的结合亲和力,因此更具靶标选择性。共价泛FGFR抑制剂(如futibatinib和resigratinib)和共价FGFR2特异性抑制剂lirafugratinib与P环中保守的半胱氨酸残基相互作用(保守残基:FGFR1为半胱氨酸488,FGFR2为半胱氨酸491,FGFR3为半胱氨酸482和FGFR4为半胱氨酸477),而共价FGFR4抑制剂(如roblitinib和fisogatinib)与FGFR4的铰链区域中的半胱氨酸552残基相互作用。非共价ATP竞争性FGFR抑制剂和共价FGFR4特异性抑制剂容易受到守门人突变的影响,这些突变妨碍药物-靶标相互作用,而共价泛FGFR或FGFR2特异性抑制剂对此类突变的敏感性较低,因此预期具有更持久的治疗效果。

图8. FGFR激酶结构域及小分子抑制剂
由Relay Therapeutics研发的RLY-4008(lirafugratinib)与FGFR2的相互作用位点主要在激酶结构域的ATP结合口袋,它通过其丙烯酰胺基团与FGFR2的Cys491形成共价键。虽然FGFR1的激酶结构域与FGFR2高度相似,但由于FGFR1的P-loop(磷酸结合环)动态性更强,RLY-4008与FGFR1的相互作用不FGFR2那么有效。FGFR1的P-loop的快速动态变化不利于RLY-4008形成稳定的共价键。与FGFR1类似,FGFR3和FGFR4的激酶结构域也与FGFR2有较高的序列和结构相似性,但在Cys491这一关键位点上没有相应的半胱氨酸残基,RLY-4008不能有效地与这些受体形成共价键。因此,RLY-4008对FGFR3和FGFR4的抑制作用远弱于对FGFR2的抑制,这使得RLY-4008具有高度的选择性。

图9. RLY-4008(lirafugratinib)结合位点示意图
近期Robert L. Hudkins等人在JMC上发表了一篇名为Discovery of TYRA-300: First Oral Selective FGFR3 Inhibitor for the Treatment of Urothelial Cancers and Achondroplasia的文章,TYRA-300是一种基于结构的强效FGFR3选择性抑制剂,治疗尿路上皮癌和软骨发育不全等疾病。TYRA-300可避免因抑制FGFR1、FGFR2和FGFR4而产生的毒性,并且不受FGFR3门卫突变影响,有潜力成为对FGFR3具有高选择性的新一代靶向疗法,为患者提供更好的抗肿瘤活性和耐受性。研究者们利用FGFR1-4激酶域的高序列和结构相似性,通过结构分析发现了FGFR3与其他FGFR亚型在铰链区域的关键差异,FGFR3在GK+4位点(守门人位点后的第四个位点)含有一个疏水性氨基酸Ala559,而FGFR1和FGFR2在相同位置分别含有极性氨基酸Ser565和Ser568。这些差异实现了TYRA-300对FGFR3的选择性抑制。


图10. TYRA-300结合位点及生化和体内药效数据
1. Javle M, King G, Spencer K, Borad MJ. Futibatinib, an Irreversible FGFR1-4 Inhibitor for the Treatment of FGFR-Aberrant Tumors. Oncologist. 2023 Nov 2;28(11):928-943.
2. Katoh M, Loriot Y, Brandi G, Tavolari S, Wainberg ZA, Katoh M. FGFR-targeted therapeutics: clinical activity, mechanisms of resistance and new directions. Nat Rev Clin Oncol. 2024 Apr;21(4):312-329.
3. Szklener K, Chmiel P, Michalski A, Mańdziuk S. New Directions and Challenges in Targeted Therapies of Advanced Bladder Cancer: The Role of FGFR Inhibitors. Cancers (Basel). 2022 Mar 10;14(6):1416.
4. Philadelphia (PA): AACR; Cancer Res 2023;83(8_Suppl):Abstract nr CT119.
5. Yue S, Li Y, Chen X, Wang J, Li M, Chen Y, Wu D. FGFR-TKI resistance in cancer: current status and perspectives. J Hematol Oncol. 2021 Feb 10;14(1):23.
6. Subbiah V, Sahai V, Maglic D, Bruderek K, Touré BB, Zhao S, Valverde R, O'Hearn PJ, Moustakas DT, Sch?nherr H, Gerami-Moayed N, Taylor AM, Hudson BM, Houde DJ, Pal D, Foster L, Gunaydin H, Ayaz P, Sharon DA, Goyal L, Schram AM, Kamath S, Sherwin CA, Schmidt-Kittler O, Jen KY, Ricard F, Wolf BB, Shaw DE, Bergstrom DA, Watters J, Casaletto JB. RLY-4008, the First Highly Selective FGFR2 Inhibitor with Activity across FGFR2 Alterations and Resistance Mutations. Cancer Discov. 2023 Sep 6;13(9):2012-2031.
7. Hudkins RL, Allen E, Balcer A, Hoffman ID, Iyer S, Neal M, Nelson KJ, Rideout M, Ye Q, Starrett JH, Patel P, Harris T, Swanson RV, Bensen DC. Discovery of TYRA-300: First Oral Selective FGFR3 Inhibitor for the Treatment of Urothelial Cancers and Achondroplasia. J Med Chem. 2024 Sep 26;67(18):16737-16756.


业务咨询
北京
业务咨询专线:010-6780-9840
联系地址:北京市经济技术开发区科创十三街18号院锋创科技园16号楼
上海
业务咨询专线:010-6780-9840
联系地址:上海市浦东新区蔡伦路780号新技术推广大楼3E5O室
徐州
业务咨询专线:010-6780-9840
联系地址:江苏省徐州市云龙区淮海文博园 二号楼2层
贵州
业务咨询专线:010-6780-9840
联系地址:贵州省贵阳市南明区龙岭路50号 欧美医药产业园一期2号楼












